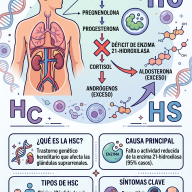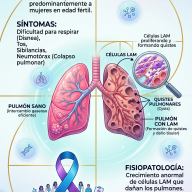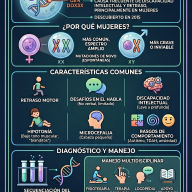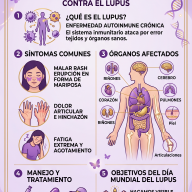
¿Qué es la Telangiectasia Hemorrágica Hereditaria o enfermedad de Rendu-Osler?
La Telangiectasia Hemorrágica Hereditaria (THH), históricamente conocida como síndrome de Osler-Weber-Rendu, es un trastorno vascular genético de carácter autosómico dominante. Aunque está catalogada dentro del grupo de las enfermedades raras debido a su prevalencia estimada de 1 caso por cada 5.000 a 8.000 personas, su impacto en la calidad de vida y el riesgo de complicaciones asociadas exigen un abordaje clínico riguroso y un conocimiento profundo por parte de la comunidad médica y de la sociedad.
A continuación, se presenta un análisis detallado sobre su fisiopatología, manifestaciones clínicas, diagnóstico y las estrategias terapéuticas actuales.
Fisiopatología: ¿Qué ocurre en el sistema vascular?
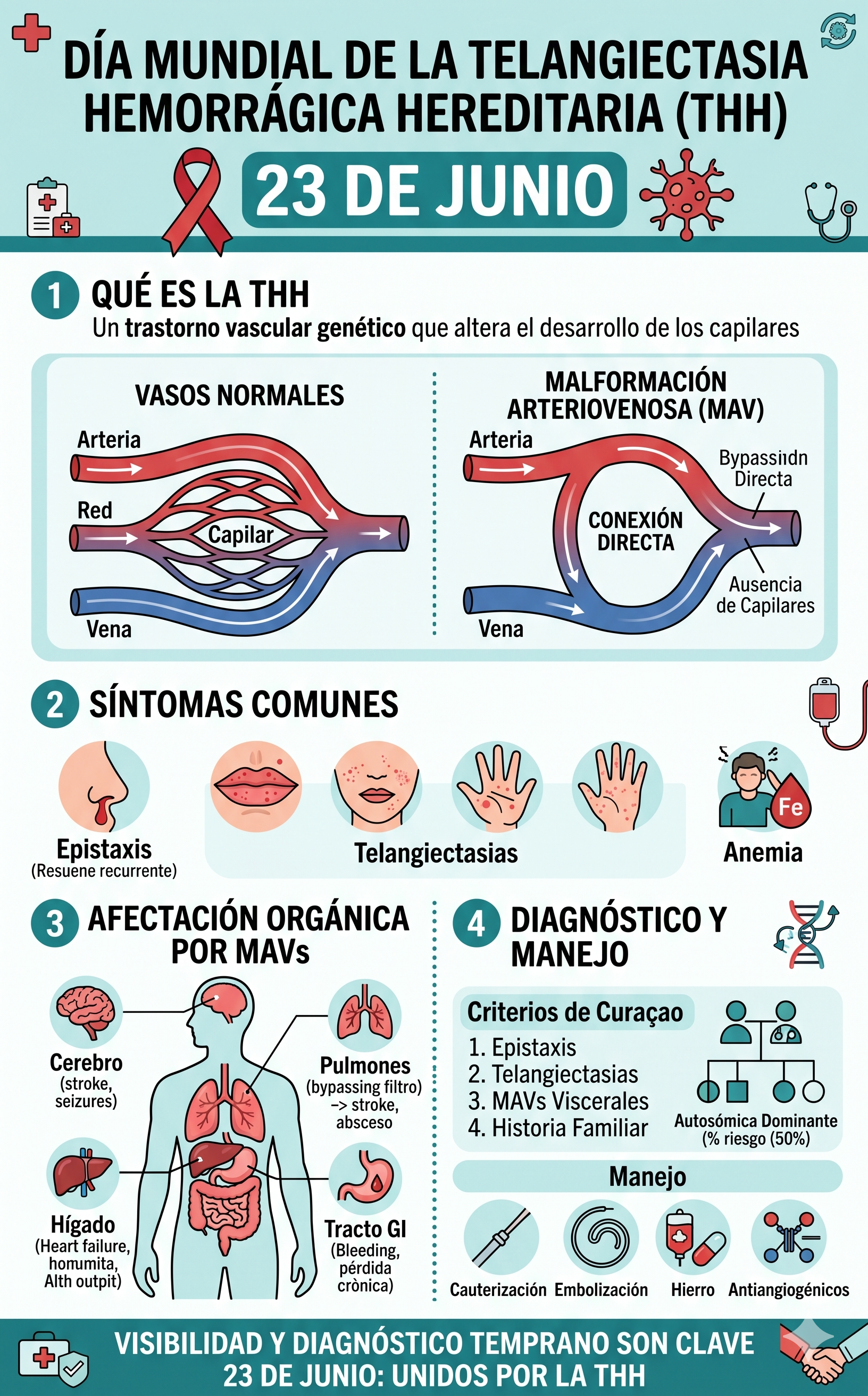
En un sistema circulatorio normal, las arterias transportan la sangre a alta presión desde el corazón hacia los órganos, ramificándose en vasos cada vez más pequeños llamados capilares. Estos capilares ralentizan el flujo sanguíneo y reducen la presión antes de que la sangre pase a las venas para regresar al corazón.
En los pacientes con THH, este mecanismo regulador falla debido a mutaciones genéticas que afectan el desarrollo de las paredes vasculares. La ausencia o disfunción de los capilares intermedios provoca que las arterias se conecten directamente con las venas. Esta alteración estructural da origen a dos tipos de lesiones características:
- Telangiectasias: Dilataciones de pequeños vasos sanguíneos cerca de la superficie de la piel o de las mucosas. Debido a la delgadez de sus paredes y a la presión interna, son extremadamente propensas a romperse y sangrar.
- Malformaciones Arteriovenosas (MAV): Conexiones anormales directas entre arterias y venas de gran calibre que se desarrollan en órganos internos como los pulmones, el cerebro o el hígado.
Como se observa en el diagrama superior, al eludirse la red capilar normal (bypassing the normal capillary network), la sangre pasa rápidamente de la arteria a la vena a una presión excesiva, lo que debilita el tejido vascular circundante y eleva el riesgo de hemorragias internas o fenómenos embólicos.
Manifestaciones Clínicas Principales
La THH es una enfermedad de expresión variable; esto significa que los síntomas y su gravedad pueden diferir considerablemente incluso entre miembros de una misma familia. Las manifestaciones más comunes incluyen:
1. Epistaxis (Sangrado nasal)
Es el síntoma inicial y más frecuente, presentándose en más del 90% de los pacientes. Suelen comenzar en la infancia o la adolescencia y se vuelven más severos y frecuentes con la edad. Pueden ocurrir de manera espontánea, incluso durante el sueño.
2. Telangiectasias Cutáneas y Mucosas
Aparecen generalmente a partir de la tercera década de la vida. Se consolidan como pequeños puntos de color rojo o violáceo localizados de forma característica en los labios, la lengua, la mucosa oral, la cara y las yemas de los dedos. Su sangrado suele ser menos severo que la epistaxis, pero puede causar incomodidad estética y funcional.
3. Afectación Orgánica Visceral (MAV internos)
Las consecuencias de las malformaciones internas dependen directamente del órgano afectado:
- Pulmonares (MAVp): Presentes en aproximadamente el 30-50% de los casos. Al no haber filtro capilar, existe el riesgo de que coágulos o bacterias pasen directamente al torrente sistémico, pudiendo ocasionar accidentes cerebrovasculares (ACV) o abscesos cerebrales. También se manifiesta mediante disnea (shortness of breath) o hipoxemia.
- Cerebrales (MAVc): Afectan a cerca del 10% de los pacientes. Aunque muchas son asintomáticas, su ruptura puede derivar en hemorragias intracraneales o convulsiones (seizures).
- Hepáticas y Gastrointestinales: Pueden provocar insuficiencia cardíaca por alto gasto (debido al exceso de sangre que procesa el hígado) o sangrados digestivos crónicos que contribuyen significativamente a la anemia por deficiencia de hierro (iron-deficiency anemia).
Genética y Patrón de Herencia
La THH se transmite de forma autosómica dominante. Esto implica que una persona afectada tiene un 50% de probabilidades de transmitir el gen mutado a cada uno de sus hijos, independientemente del sexo.
A nivel molecular, la enfermedad está vinculada principalmente a mutaciones en tres genes implicados en la vía de señalización del factor de crecimiento transformante beta (TGF-β), esencial para la angiogénesis (formación de vasos sanguíneos):
| Gen Mutado | Tipo de THH | Características Predominantes |
| ENG (Endoglina) | THH Tipo 1 | Mayor incidencia de Malformaciones Arteriovenosas pulmonares y cerebrales. |
| ACVRL1 (ALK1) | THH Tipo 2 | Mayor propensión a la afectación hepática y diagnóstico más tardío. |
| SMAD4 | Síndrome de superposición | Combina síntomas de THH con poliposis juvenil (riesgo de pólipos gastrointestinales). |
Criterios de Diagnóstico: Los Criterios de Curaçao
En la práctica clínica, el diagnóstico de la Telangiectasia Hemorrágica Hereditaria se establece a partir de directrices internacionales conocidas como los Criterios de Curaçao.
Un paciente recibe el diagnóstico definitivo de THH si cumple con al menos tres de los siguientes cuatro criterios:
- Epistaxis: Sangrados nasales espontáneos y recurrentes.
- Telangiectasias: Múltiples y en localizaciones características (labios, cavidad oral, dedos, nariz).
- Afectación visceral: Demostración de MAV en pulmón, hígado, cerebro o tracto digestivo.
- Historia familiar: Un pariente de primer grado diagnosticado formalmente con la enfermedad.
Si se cumplen únicamente dos criterios, el diagnóstico se considera sospechoso o posible, requiriendo confirmación mediante estudios genéticos moleculares.
Opciones de Tratamiento y Manejo Clínico
En la actualidad, la THH no cuenta con una cura definitiva. El enfoque médico es fundamentalmente sintomático, preventivo y multidisciplinar, orientado a mitigar los sangrados y evitar complicaciones graves derivados de las MAV.
Manejo de la Epistaxis y la Anemia
- Tópico e Hidratación: Uso de pomadas lubricantes o soluciones salinas para mantener la mucosa nasal húmeda y reducir la frecuencia del sangrado.
- Procedimientos Locales: Ablación con láser o cauterización química de las telangiectasias nasales.
- Tratamiento de la Anemia: Administración de suplementos de hierro por vía oral o intravenosa. En casos de sangrado masivo, se recurre a transfusiones de sangre.
Tratamiento de Malformaciones Internas
- Embolización por Cateterismo: Es el tratamiento de elección para las MAV pulmonares y cerebrales accesibles. Consiste en obstruir de forma selectiva los vasos anómalos mediante la introducción de pequeñas espirales metálicas (coils) o sustancias oclusivas para eliminar el riesgo de ruptura y filtración.
Avances Farmacológicos (Terapias Antiangiogénicas)
En los últimos años, el uso de fármacos que inhiben la formación descontrolada de vasos sanguíneos ha supuesto una revolución en el manejo de casos graves. Medicamentos como el bevacizumab (un anticuerpo monoclonal que bloquea el factor de crecimiento endotelial vascular) han demostrado una alta eficacia reduciendo drásticamente la frecuencia de las epistaxis y estabilizando el gasto cardíaco derivado de la afectación hepática.
últimas noticias sobre Enfermedad de Rendu-Osler-Weber