Asociación Española de Hiperplasia Suprarrenal Congénita
1. Introducción y Definición
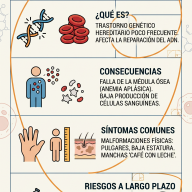
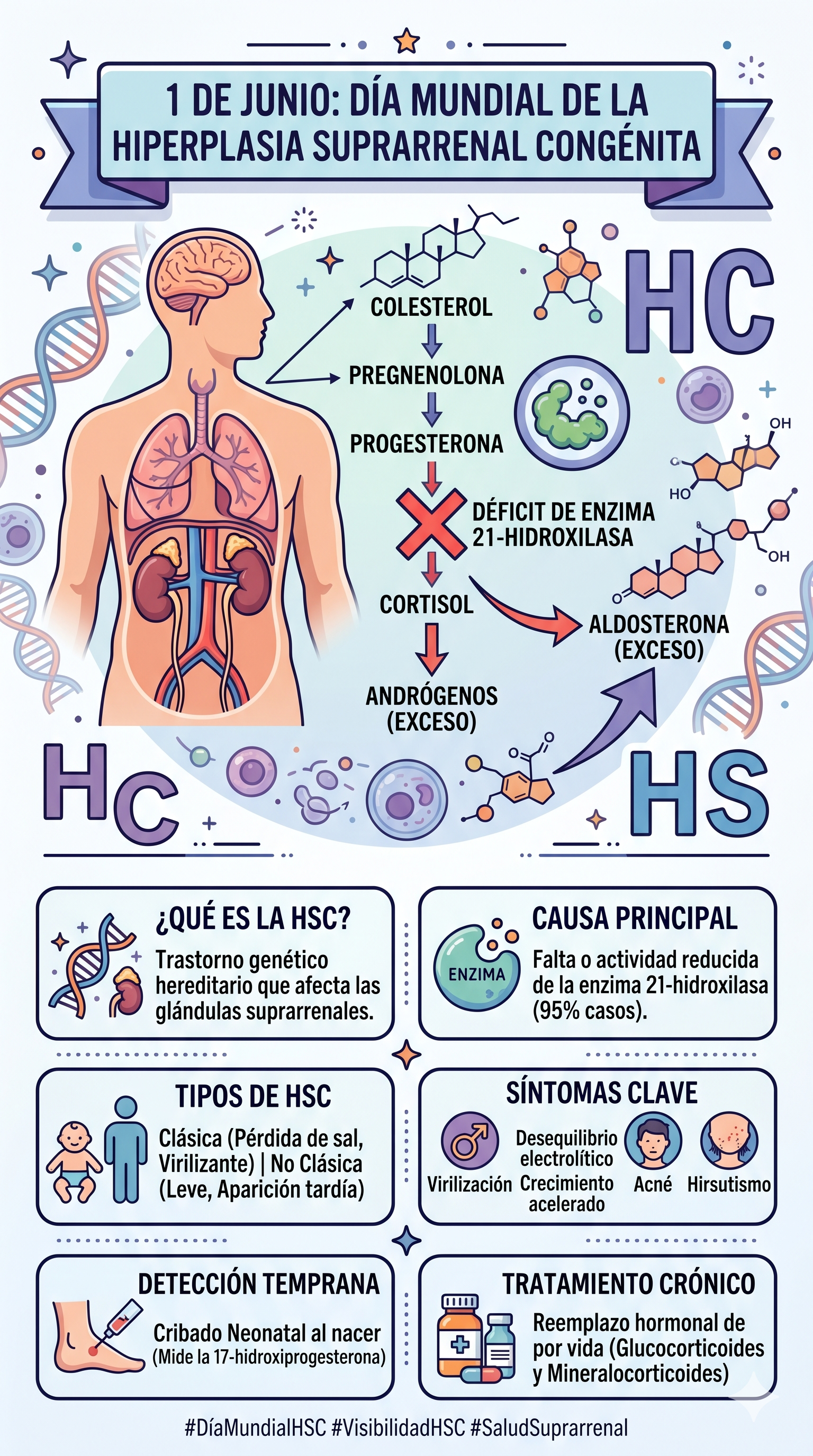
La Hiperplasia Suprarrenal Congénita (HSC) comprende un grupo de trastornos genéticos hereditarios de naturaleza autosómica recesiva. Esta condición impacta directamente la esteroidogénesis en las glándulas suprarrenales —estructuras endocrinas ubicadas en el polo superior de ambos riñones—.
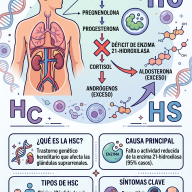
En los individuos afectados, la presencia de mutaciones genéticas específicas se traduce en la deficiencia funcional de alguna de las enzimas esenciales para la síntesis de hormonas esteroideas. El déficit enzimático genera un bloqueo o "embotellamiento" metabólico en la corteza suprarrenal. Al interrumpirse la ruta hacia la síntesis de glucocorticoides y mineralocorticoides, los precursores acumulados se desvían de manera masiva hacia la ruta biosintética de los andrógenos (hormonas sexuales masculinas).
El defecto molecular más prevalente —responsable de aproximadamente el 95% de los casos clínicos— ocurre en la enzima 21-hidroxilasa. Como se ilustra en el diagrama metabólico, el fallo en este punto crítico impide la transformación de la 17-hidroxiprogesterona (17-OHP) en cortisol y de la progesterona en aldosterona, gatillando una hiperproducción de testosterona y otros compuestos virilizantes.
2. Genética y Mecanismo de Herencia
La HSC se transmite bajo un patrón de herencia autosómico recesivo. Para que la patología se manifieste clínicamente, el paciente debe heredar dos alelos mutados del gen afectado (un alelo de procedencia materna y otro de procedencia paterna).
Cuando ambos progenitores actúan como portadores heterocigotos asintomáticos (poseen un alelo mutado y un alelo funcional), la probabilidad estadística en cada gestación se distribuye de la siguiente manera:
- 25% de probabilidad: Individuo homocigoto afectado (manifiesta la enfermedad).
- 50% de probabilidad: Individuo heterocigoto portador asintomático (transmite el alelo mutado pero no desarrolla la patología).
- 25% de probabilidad: Individuo homocigoto sano (no afectado y no portador).
El gen implicado en la gran mayoría de los casos es el CYP21A2, mapeado en el cromosoma 6p21.3. La severidad del cuadro clínico guarda una correlación directa con el tipo de mutación molecular; las deleciones completas o mutaciones de empalme (splicing) graves anulan por completo la actividad enzimática, mientras que las mutaciones puntuales o de sentido erróneo (missense) suelen conservar cierta actividad residual, modulando la gravedad del fenotipo.
3. Clasificación Clínica y Sintomatología
La presentación clínica de la HSC es heterogénea y se categoriza en dos variantes principales según el grado de función enzimática residual:
3.1. HSC Clásica (Forma Grave)
Se evidencia desde el nacimiento o durante las primeras semanas de vida. Se subdivide en:
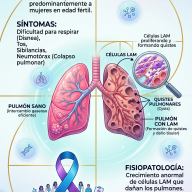
- Forma perdedora de sal: Representa el escenario de mayor compromiso vital. La ausencia de actividad de la 21-hidroxilasa es absoluta, lo que anula la producción tanto de cortisol como de aldosterona. La carencia de aldosterona impide la reabsorción de sodio en los túbulos renales, desencadenando una excreción masiva de sal y agua. Clínicamente se manifiesta de forma aguda en los neonatos mediante vómitos, pérdida ponderal acelerada, deshidratación profunda, hiponatremia, hiperpotasemia, hipotensión y shock hipovolémico (crisis suprarrenal).
- Forma virilizante simple: Existe una actividad enzimática residual mínima (1-2%) suficiente para mantener una homeostasis hidroelectrolítica aceptable mediante la secreción de cantidades basales de aldosterona. No obstante, el déficit de cortisol induce una elevación compensatoria de la hormona adrenocorticotropa (ACTH) por retroalimentación negativa, cronificando la sobreproducción de andrógenos suprarrenales.
3.2. HSC No Clásica (Forma Leve o de Aparición Tardía)
Es la variante más frecuente y benigna. Cuenta con una actividad enzimática del 20% al 50%. Los pacientes nacen sin anomalías estructurales y no experimentan crisis de pérdida de sal. El cuadro clínico debuta de forma tardía (infancia tardía, adolescencia o adultez temprana) provocado por el hiperandrogenismo periférico moderado:
- Manifestaciones generales: Aceleración de la velocidad de crecimiento en la infancia asociada a una maduración ósea precoz, lo que paradójicamente causa una talla baja final al cerrarse antes de tiempo las epífisis de los huesos largos. Acné quístico refractario a tratamientos convencionales.
- En mujeres: Hirsutismo (exceso de vello terminal en zonas dependientes de andrógenos), irregularidades en el ciclo menstrual (oligomenorrea o amenorrea), síndrome de ovarios poliquísticos secundario y subfertilidad.
Summary de sintomatología según sexo biológico en la forma clásica:
- Recién nacidas (46,XX): La exposición a concentraciones elevadas de andrógenos intrauterinos provoca la virilización de los genitales externos (hipertrofia del clítoris, fusión labioescrotal), resultando en genitales ambiguos al nacer. Los órganos derivados de los conductos de Müller (útero, trompas de Falopio y vagina proximal) permanecen intactos y sanos.
- Recién nacidos (46,XY): Presentan una anatomía genital externa fenotípicamente normal. Su diagnóstico en ausencia de cribado neonatal suele retrasarse hasta la aparición de una crisis perdedora de sal en las primeras semanas de vida, o bien durante la infancia temprana por signos de pubertad precoz isosexual.
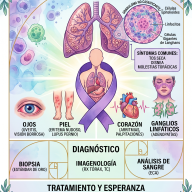
4. Protocolo Diagnóstico Avanzado
El abordaje diagnóstico combina el tamizaje poblacional, pruebas funcionales endocrinas y herramientas de biología molecular:
- Cribado Neonatal (Tamiz): Consiste en la cuantificación de los niveles de 17-hidroxiprogesterona (17-OHP) a partir de muestras de sangre capilar obtenidas en papel filtro (prueba del talón) entre las 48 y 72 horas de vida. Concentraciones drásticamente elevadas de este marcador confirman el diagnóstico presuntivo de la forma clásica.
- Prueba de Estimulación con ACTH: Constituye el estándar de oro para el diagnóstico de la forma no clásica. Se administra una dosis endovenosa de cosintropina (ACTH sintética) y se determinan los niveles basales y post-estímulo (a los 60 minutos) de la 17-OHP. Una respuesta de elevación exponencial traza el diagnóstico definitivo.
- Genotipado Molecular: Secuenciación directa o análisis de amplificación de sondas dependiente de ligandos múltiples (MLPA) del gen CYP21A2. Permite mapear las mutaciones exactas, establecer una correlación genotipo-fenotipo precisa y guiar el consejo genético reproductivo para la familia.
5. Esquema de Tratamiento y Seguimiento Clínico
El manejo de la HSC es crónico, multidisciplinario y de alta precisión, enfocado en sustituir los déficits hormonales y suprimir la vía androgénica excesiva.
5.1. Terapia de Reemplazo Farmacológico
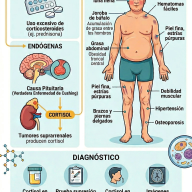
- Glucocorticoides: Sustituyen la falta de cortisol y actúan frenando la secreción hipofisaria de ACTH, reduciendo indirectamente la síntesis de andrógenos. En pacientes pediátricos se prefiere el uso de hidrocortisona por su vida media corta y menor impacto deletéreo en el crecimiento lineal. En pacientes adultos con maduración ósea completada, se pueden emplear glucocorticoides de acción prolongada como la prednisona o la dexametasona.
- Mineralocorticoides: Indispensables en la forma perdedora de sal. Se prescribe fludrocortisona por vía oral para normalizar la reabsorción de sodio, regular los niveles de potasio sérico y estabilizar la presión arterial.
- Suplementación de Cloruro de Sodio: Se añade sal suplementaria a la dieta o fórmulas lácteas durante el primer año de vida en lactantes con la forma perdedora de sal, debido a la inmadurez renal fisiológica para retener el ion sodio.
⚠️ Protocolo de Estrés Físico (Pauta de Emergencia): Los pacientes con HSC clásica carecen de la capacidad de secretar cortisol endógeno adaptativo ante agresiones biológicas. Ante eventos de estrés fisiológico moderado a grave (fiebre mayor a 38.5°C, infecciones sistémicas, fracturas, gastroenteritis con intolerancia a la vía oral o procedimientos quirúrgicos), se debe duplicar o triplicar la dosis habitual de glucocorticoides por vía oral, o administrar hidrocortisona por vía intramuscular/endovenosa de forma inmediata para evitar una crisis suprarrenal letal.
5.2. Monitoreo Terapéutico y Complicaciones
El mantenimiento de la dosificación idónea es complejo y requiere un estrecho balance bioquímico y clínico:
| Vector de Evaluación | Consecuencias de la Subdosificación (Deficiencia terapéutica) | Consecuencias de la Sobredosificación (Exceso terapéutico) |
| Desarrollo Óseo | Avance acelerado de la edad ósea sobre la cronológica; fusión epifisaria temprana con compromiso severo de la talla adulta. | Supresión directa del crecimiento longitudinal; inducción de desmineralización ósea (osteopenia/osteoporosis precoz). |
| Metabolismo | Fatiga, letargo, tendencia a la hipoglucemia y vulnerabilidad a crisis suprarrenales agudas. | Aparición de un fenotipo Cushingoide: ganancia de peso central, estrías purpúreas, resistencia a la insulina e hipertensión arterial. |
| Eje Gonadal |
Mujeres: Virilización progresiva, hirsutismo severo, anovulación crónica e infertilidad. Varones: Desarrollo de tumores de restos suprarrenales testiculares (TART) por estimulación crónica de ACTH, que pueden inducir dolor local y azoospermia obstructiva. |
Supresión del eje hipotálamo-hipófisis-gonadal por acción directa del exceso de esteroides exógenos, alterando la pulsatilidad de las gonadotropinas y comprometiendo igualmente la fertilidad. |
6. Perspectivas de Investigación y Terapias Emergentes
La evolución médica busca mitigar la exposición crónica a dosis suprafisiológicas de glucocorticoides mediante el desarrollo de dianas terapéuticas más selectivas:
- Glucocorticoides de Liberación Modificada: Formulaciones de hidrocortisona diseñadas con una cinética de liberación retardada que mimetizan el ritmo circadiano fisiológico del cortisol (pico máximo en el periodo pre-despertar). Esto optimiza la supresión nocturna de ACTH y reduce la carga total diaria de esteroides requerida.
- Moduladores del Eje Neuroendocrino (Antagonistas del Receptor de CRH): Fármacos actualmente bajo ensayos clínicos avanzados que bloquean de forma competitiva los receptores de la hormona liberadora de corticotropina (CRH) en la hipófisis. Al atenuar el estímulo central que orquesta la secreción de ACTH, viabilizan un control eficaz del exceso de andrógenos utilizando dosis de corticoides significativamente menores, disminuyendo drásticamente el riesgo de comorbilidades metabólicas a largo plazo.
últimas noticias sobre Hiperplasia suprarrenal congénita
Últimas noticias