SYNGAP1 Global Network (SGN) | SYNGAP1 Argentina
El trastorno relacionado con el gen SYNGAP1 es una condición del neurodesarrollo rara y compleja causada por una mutación o variante en el gen SYNGAP1, ubicado en el cromosoma 6. Debido al papel crítico que juega este gen en la comunicación cerebral, su alteración desencadena un espectro de desafíos neurológicos, cognitivos, físicos y conductuales que impactan profundamente a quienes lo padecen y a sus familias.
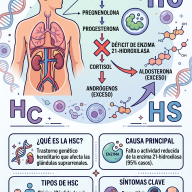
🧬 La Biología detrás de SYNGAP1: ¿Qué falla en el cerebro?
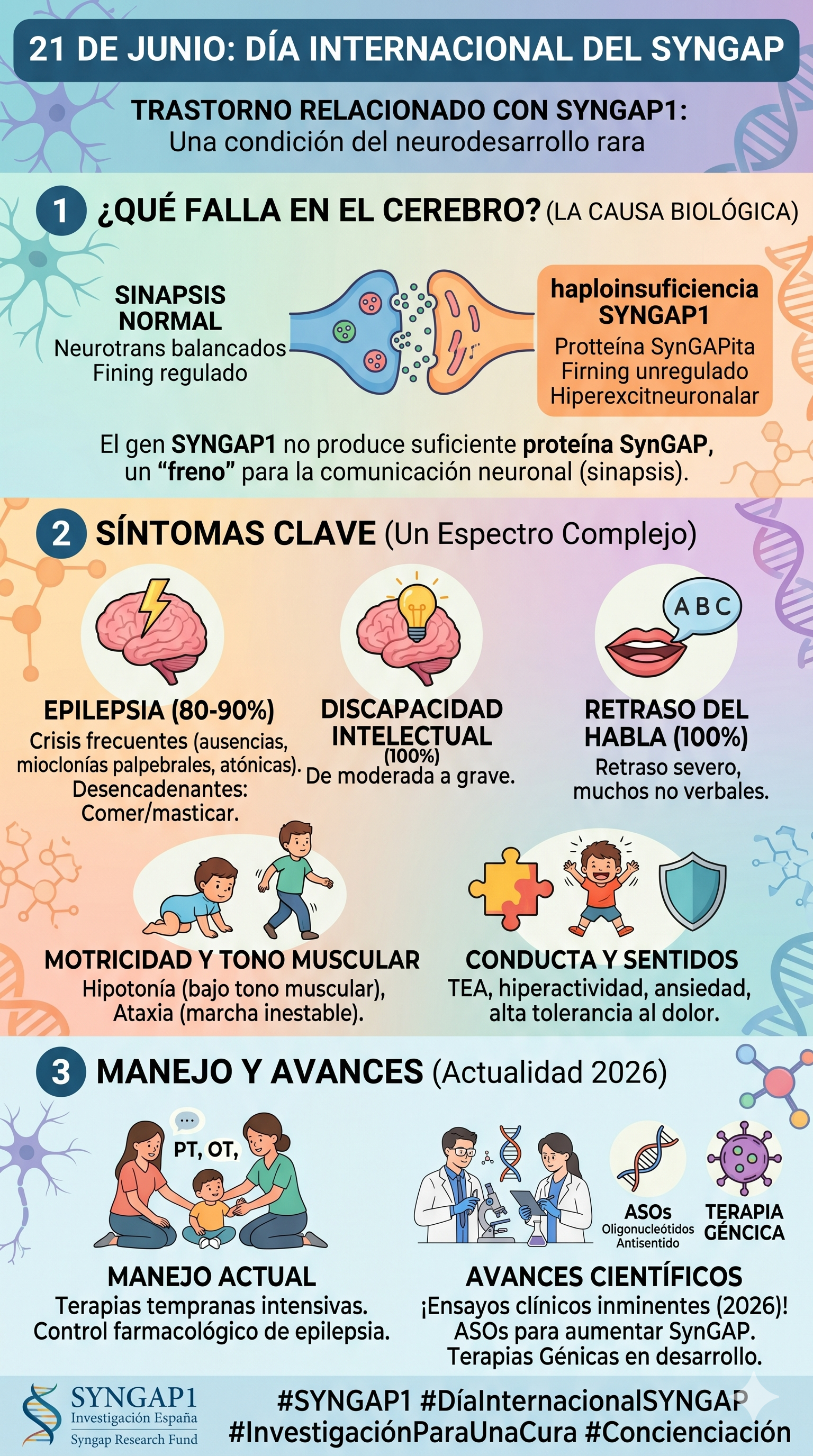
Para entender el trastorno, primero debemos comprender la función del gen afectado. El gen SYNGAP1 contiene las instrucciones necesarias para fabricar la proteína SynGAP. Esta proteína se concentra intensamente en las sinapsis, los puntos de contacto y comunicación química entre las neuronas.
- El regulador del aprendizaje: La proteína SynGAP actúa como un "freno" o regulador crucial en el sistema nervioso. Controla la plasticidad sináptica, que es la capacidad del cerebro para cambiar, adaptarse, memorizar y procesar los estímulos del entorno.
- Haploinsuficiencia: La inmensa mayoría de los pacientes presentan lo que en genética se denomina haploinsuficiencia. Tienen una copia del gen sana y otra dañada. Como resultado, el organismo solo produce aproximadamente el 50% de la proteína SynGAP que necesita. Sin el "freno" suficiente de esta proteína, los circuitos cerebrales experimentan una actividad excesiva o hiperexcitabilidad.
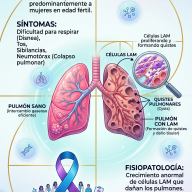
📋 Síntomas y Características Clínicas
El SYNGAP1 es un trastorno de espectro, lo que significa que la gravedad y la combinación de los síntomas varían de una persona a otra. Sin embargo, existen tres pilares clínicos fundamentales:
1. Discapacidad Intelectual y Retrasos del Desarrollo
- Déficit cognitivo: Afecta prácticamente al 100% de los pacientes, manifestándose generalmente como una discapacidad intelectual que va de moderada a grave.
- Hitos motores retrasados: Existe una notable dificultad para alcanzar metas físicas básicas como sentarse, gatear o caminar. Es habitual observar una marcha inestable conocida como ataxia.
- Compromiso del habla: El retraso en el lenguaje es severo; un porcentaje considerable de los pacientes permanece en una condición no verbal o requiere sistemas aumentativos y alternativos de comunicación (SAAC).
2. Epilepsia y Crisis Convulsivas
- Afecta a un rango estimado de entre el 80% y el 90% de las personas diagnosticadas, con un inicio que suele ocurrir durante la primera infancia.
- Las crisis pueden ser masivas y recurrentes (hasta docenas o cientos al día). Destacan las crisis de ausencia (desconexión breve con mirada fija), las mioclonías palpebrales (parpadeo involuntario e incontrolable) y las crisis atónicas (caídas abruptas por pérdida repentina del tono muscular).
- Desencadenante sensorial específico: Un rasgo sumamente característico en cerca del 25% de los casos es la crisis refleja por ingesta, donde el acto de comer o masticar activa las convulsiones.
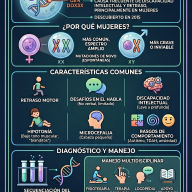
3. Rasgos Conductuales y Sensoriales
- Autismo: Aproximadamente la mitad de los afectados recibe un diagnóstico comórbido de Trastorno del Espectro Autista (TEA).
- Conducta: Son comunes la hiperactividad, la impulsividad, la ansiedad severa, los trastornos del sueño y las crisis de agresividad o conductas autolesivas.
- Hipotonía: El tono muscular bajo o blando es una constante que dificulta la fuerza y la motricidad.
- Alta tolerancia al dolor: Un número elevado de pacientes muestra un umbral del dolor inusualmente alto, lo que puede enmascarar lesiones físicas o enfermedades médicas subyacentes.
🔍 Diagnóstico y Origen Genético
Dado que los síntomas del SYNGAP1 coinciden con muchas otras afecciones del neurodesarrollo, el diagnóstico clínico es imposible sin herramientas avanzadas. La confirmación médica se realiza exclusivamente a través de pruebas genéticas, tales como:
- Secuenciación del Exoma Completo (WES).
- Paneles genéticos dirigidos específicamente a la epilepsia o al autismo.
En la gran mayoría de los casos detectados, la mutación es de tipo de novo. Esto significa que la alteración genética ocurrió de forma espontánea durante la formación del óvulo o el espermatozoide, o bien en las primeras etapas del desarrollo embrionario; no se hereda de los padres ni aumenta significativamente el riesgo de que se repita en futuros embarazos de la pareja.
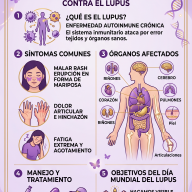
💊 Tratamientos Estándar y Manejo Clínico
A día de hoy, no existe una cura definitiva, pero el tratamiento multidisciplinar temprano resulta decisivo para mejorar la autonomía y calidad de vida del paciente:
- Terapias de apoyo intensivas: Fisioterapia (para la hipotonía y la marcha), Terapia Ocupacional (para la integración sensorial y motricidad fina), Logopedia (para potenciar la comunicación) y terapias conductuales especializadas como el Análisis Conductual Aplicado (ABA).
- Control farmacológico de la epilepsia: Se recurre a una amplia gama de medicamentos anticonvulsivos. Sin embargo, la epilepsia en SYNGAP1 suele ser farmacorresistente. En estos escenarios se implementan terapias alternativas como la dieta cetogénica médica o la implantación de un Estimulador del Nervio Vago (VNS).
🚀 Avances Recientes en Investigación (Actualidad 2026)
El enfoque de la medicina para SYNGAP1 ha dado un giro histórico, transitando de los cuidados puramente paliativos al desarrollo de terapias moleculares dirigidas a corregir la raíz biológica del problema. Los dos campos más prometedores son:
1. Oligonucleótidos Antisentido (ASO)
Los ASO son cadenas cortas y sintéticas de nucleótidos que se administran periódicamente en el líquido cefalorraquídeo mediante inyección intratecal. Su objetivo es unirse al ARN para modificar la expresión génica y forzar a la copia sana del gen a producir más proteína.
- El fármaco CMP-002 (CAMP4 Therapeutics): Se consolida como uno de los proyectos más avanzados. Tras presentar datos preclínicos robustos que demostraron un aumento de la proteína SynGAP, una reducción drástica de las convulsiones y mejoras en el comportamiento en modelos animales, la compañía ha confirmado el inicio de su ensayo clínico de Fase 1/2 en humanos para la segunda mitad de 2026.
- Plataformas de reactivación (Stoke Therapeutics / Acadia): Utilizando el éxito previo de plataformas de ASO implementadas en otras encefalopatías epilépticas (como el Síndrome de Dravet), estos laboratorios optimizan moléculas candidatas en células madre de pacientes para estandarizar dosis seguras de cara a futuros ensayos.
2. Terapia Génica y Vectores Adaptados
La terapia génica aspira a ser una solución definitiva de una sola aplicación introduciendo una copia completamente funcional del gen en las neuronas a través de vectores de virus modificados (AAV).
- El reto del tamaño: El gen SYNGAP1 es demasiado grande para los vectores de transporte tradicionales. Para superar este obstáculo físico, las investigaciones recientes se han enfocado en el uso de isoformas específicas (se seleccionan únicamente los fragmentos esenciales del gen que regulan la plasticidad y la epilepsia, reduciendo su tamaño físico) y en el diseño de vectores de gran capacidad. Los ensayos en modelos preclínicos confirman que estas técnicas logran rescatar eficazmente las funciones cognitivas y restaurar la normalidad eléctrica de las sinapsis.
💡 Conclusión
El trastorno relacionado con SYNGAP1 se encuentra en un punto de inflexión. El esfuerzo coordinado entre las organizaciones internacionales de pacientes, que impulsan registros globales de historia natural y la búsqueda de biomarcadores, y las compañías biotecnológicas ha acelerado los tiempos de desarrollo. El paso de los ensayos de laboratorio a las pruebas clínicas en humanos abre una ventana de esperanza histórica para transformar el tratamiento de esta enfermedad.
últimas noticias sobre SYNGAP
Últimas noticias