La Esclerosis Lateral Amiotrófica (ELA) es una enfermedad neurodegenerativa progresiva que afecta al sistema nervioso central, específicamente a las neuronas encargadas de controlar el movimiento muscular voluntario. Es una de las patologías neuromusculares más complejas de la medicina actual.
1. Fisiopatología: ¿Qué ocurre en el sistema nervioso?
El origen de la ELA radica en la muerte celular de las motoneuronas (las células nerviosas que transmiten las órdenes motrices desde el cerebro hasta los músculos). En esta enfermedad, se ven afectados simultáneamente dos niveles del sistema motor:
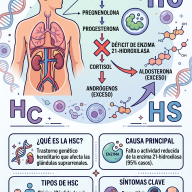
- Motoneuronas superiores (en la corteza cerebral): Su degeneración interrumpe la primera parte de la señal eléctrica. Esto provoca espasticidad (rigidez y tensión muscular excesiva) y reflejos anormalmente exaltados (hiperreflexia).
- Motoneuronas inferiores (en el tronco del encéfalo y la médula espinal): Son las que conectan directamente con los músculos. Su pérdida causa atrofia (pérdida de masa muscular), debilidad extrema y fasciculaciones (pequeños espasmos voluntarios perceptibles bajo la piel).
A nivel celular, este proceso se produce por una combinación de factores: acumulación de proteínas defectuosas (como la TDP-43), estrés oxidativo, inflamación del tejido nervioso y disfunción de las mitocondrias (las centrales de energía de las células), lo que aboca a la neurona a su destrucción.
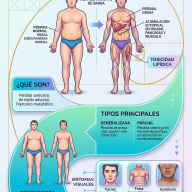
2. Clasificación de la ELA
La enfermedad presenta variaciones según su origen biológico y la región del cuerpo donde se manifiestan los primeros síntomas.
Según su origen genético
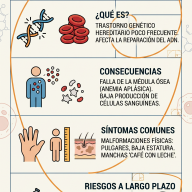
- ELA Esporádica (90-95% de los casos): Es la forma más común. Aparece de forma aleatoria, sin antecedentes familiares previos. Se considera el resultado de una interacción compleja entre predisposición genética y factores ambientales aún no determinados.
- ELA Familiar (5-10% de los casos): Se presenta cuando existen dos o más miembros afectados en una misma línea genética. Se debe a mutaciones hereditarias directas en genes específicos, siendo los más estudiados el C9orf72, SOD1, TARDBP y FUS.
Según el inicio de los síntomas
La zona donde mueren las primeras motoneuronas determina cómo debuta la enfermedad:
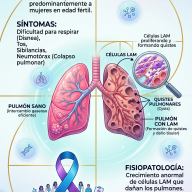
- Inicio Espinal (70-75% de los casos): Los síntomas comienzan en las extremidades. El paciente experimenta debilidad en un brazo o una pierna, caídas sutiles, dificultad para girar una llave, abrocharse los botones o levantar objetos livianos.
- Inicio Bulbar (25-30% de los casos): Afecta inicialmente a las neuronas del tronco encefálico que controlan los músculos de la boca y la garganta. Se manifiesta con dificultad para pronunciar palabras (disartria), problemas para tragar alimentos o líquidos (disfagia) o cambios en el tono de la voz.
3. Fases de progresión de la enfermedad
Aunque la velocidad de evolución es estrictamente individual, la enfermedad avanza de forma progresiva a través de las siguientes etapas clínicas:
- Fase Inicial (Debut Localizado): Los síntomas se limitan a una única región corporal (por ejemplo, una mano o el habla). El paciente mantiene un alto nivel de independencia, aunque experimenta fatiga y pérdida leve de destreza en sus actividades diarias.
- Fase Intermedia (Diseminación Multirregional): La debilidad se extiende a otras áreas del cuerpo. La autonomía se reduce significativamente, requiriendo el uso de ayudas técnicas como bastones, sillas de ruedas o sistemas de comunicación adaptada. Los problemas de deglución exigen modificar la textura de la comida para evitar atragantamientos.
- Fase Avanzada (Dependencia Total y Soporte Vital): La parálisis muscular afecta a casi la totalidad del cuerpo, incluidos los músculos respiratorios (el diafragma). El paciente requiere asistencia continua. Las funciones cognitivas, la sensibilidad táctil, la vista, el oído y el control de los esfínteres suelen mantenerse intactos.
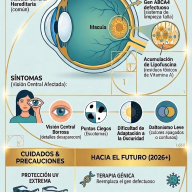
4. El proceso diagnóstico
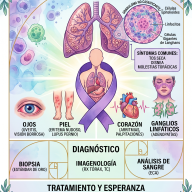
No existe un análisis de sangre o una prueba de imagen única que dictamine con total certeza que un paciente padece ELA. El diagnóstico se realiza por exclusión, descartando otras patologías neurológicas tratables (como la esclerosis múltiple, neuropatías periféricas o compresiones de la médula espinal).
Los neurólogos se basan en tres pilares fundamentales:
- Exploración clínica neurológica: Evidenciar signos combinados de afectación de motoneurona superior e inferior en múltiples regiones corporales.
- Electromiografía (EMG): Prueba crucial que mide la actividad eléctrica de los músculos. Permite detectar la pérdida de conexión nerviosa (denervación) incluso en músculos que clínicamente aún parecen fuertes.
- Resonancia Magnética (RMN) y analíticas: Utilizadas para descartar tumores, hernias discales, infecciones, déficits vitamínicos graves o trastornos autoinmunes.
5. Abordaje terapéutico actual
En la actualidad, la ELA no tiene una cura que revierta el daño neuronal, pero la medicina dispone de tratamientos diseñados para ralentizar la progresión, controlar los síntomas y mejorar la calidad de vida del paciente a través de un enfoque multidisciplinar.
Tratamiento Farmacológico
- Moduladores de la enfermedad: Medicamentos como el Riluzol actúan reduciendo la acumulación de glutamato (un neurotransmisor que en exceso destruye las neuronas), logrando prolongar la supervivencia y ralentizar discretamente el avance de la patología.
- Tratamiento de síntomas: Se emplean fármacos específicos para paliar el dolor, los calambres musculares, la rigidez, el exceso de saliva (sialorrea) y la labilidad emocional (episodios involuntarios de risa o llanto).
Soporte Técnico e Intervenciones Clave
El cuidado diario se centra en compensar la pérdida de función muscular mediante procedimientos especializados:
- Soporte Respiratorio: La aplicación de ventilación no invasiva (VNI) mediante mascarillas conectadas a un ventilador portátil estabiliza la respiración, especialmente por las noches, aliviando el cansancio y el dolor de cabeza matutino.
- Soporte Nutricional: Cuando la disfagia es severa, se realiza la colocación de una Gastrostomía Endoscópica Percutánea (PEG), un pequeño tubo directo al estómago que garantiza la hidratación y nutrición correctas, evitando desnutrición e infecciones pulmonares por aspiración.
- Equipos Multidisciplinares: La intervención coordinada de fisioterapeutas (mantenimiento de elasticidad), terapeutas ocupacionales (adaptación del hogar), psicólogos y logopedas (entrenamiento en sistemas de comunicación alternativa mediante seguimiento ocular) resulta indispensable para el bienestar del paciente y su entorno familiar.
últimas noticias sobre Esclerosis Lateral Amiotrófica
Últimas noticias