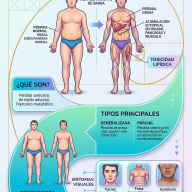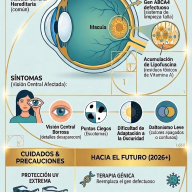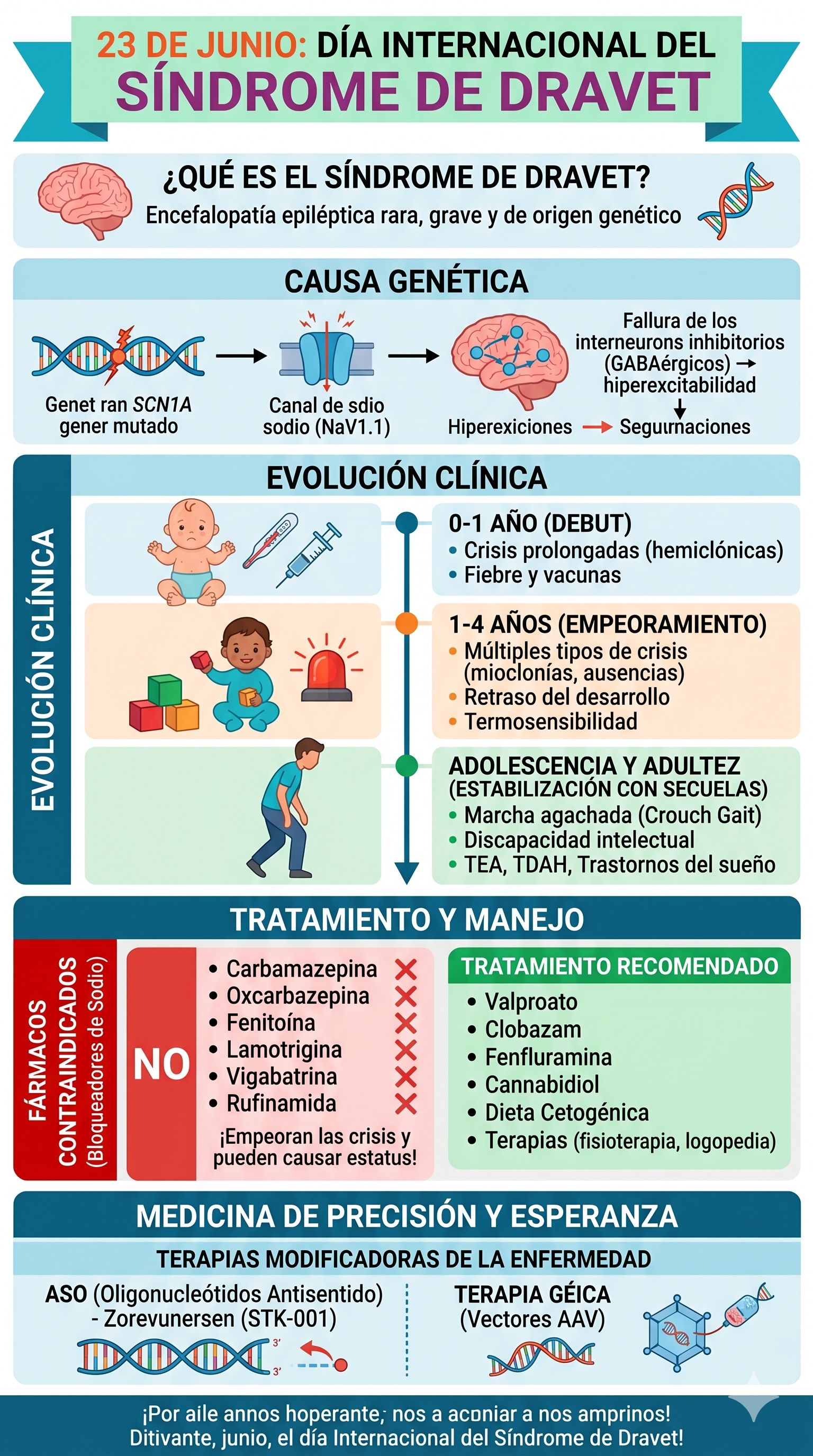
El Síndrome de Dravet, antiguamente conocido como Epilepsia Mioclónica Severa de la Infancia (EMSI), es una variante rara y muy grave de epilepsia de origen genético. Está clasificado dentro de las encefalopatías epilépticas y del desarrollo, un término que indica que tanto la actividad epiléptica continua como la anomalía genética subyacente contribuyen activamente al retraso progresivo en las áreas cognitiva, motora y conductual del paciente.
A continuación, se presenta el cuerpo completo de información clínica, estructurado para su consulta, archivo o reproducción íntegra.
1. Etiología y Mecanismo Neurobiológico
En aproximadamente el 80-85% de los casos, el Síndrome de Dravet es causado por una mutación puntual o una deleción en el gen SCN1A, ubicado en el cromosoma 2q24.
- Mutaciones De Novo: En la gran mayoría de los pacientes, estas mutaciones aparecen de forma espontánea (mutación de novo), lo que significa que los padres no son portadores ni padecen la enfermedad.
- El Canal de Sodio $Na_V1.1$: El gen SCN1A codifica la subunidad alfa-1 del canal de sodio dependiente de voltaje Na_V1.1, indispensable para la transmisión de impulsos eléctricos en el sistema nervioso central.
- Fallo en la Inhibición Cerebral (Haploinsuficiencia): La mutación provoca que una de las dos copias del gen no funcione correctamente. Esto afecta de manera selectiva a las interneuronas GABAérgicas (las neuronas encargadas de frenar y regular la actividad eléctrica cerebral). Al perderse este "freno" natural, el cerebro queda desprotegido ante estímulos comunes, desencadenando crisis epilépticas por hiperexcitabilidad.
2. Evolución Clínica y Síntomas por Etapas
El debut de la enfermedad altera por completo el desarrollo de un lactante que, hasta ese momento, mostraba un crecimiento y unos hitos madurativos perfectamente normales.
Primer Año de Vida (Fase de Debut)
- La Primera Crisis: Aparece generalmente entre los 3 y los 9 meses de edad.
- Desencadenantes Térmicos: El principal detonante es la elevación de la temperatura corporal, ya sea por fiebre (asociada a infecciones leves) o de forma secundaria tras la administración de vacunas de la infancia. Las vacunas no causan la enfermedad, actúan únicamente como un desencadenante térmico.
- Tipología: Las crisis iniciales suelen ser prolongadas, superando con frecuencia los 15 o 20 minutos (evolucionando a status epilepticus o estado de mal epiléptico). Tienden a ser de carácter hemiclónico (sacudidas que afectan a una sola mitad del cuerpo) y tienen la particularidad de alternar de lado de un episodio a otro.
De 1 a 4 Años (Fase de Empeoramiento)
- Diversificación de las Crisis: Las crisis térmicas persisten, pero aparecen nuevos tipos de crisis sin necesidad de fiebre: mioclonías (sacudidas musculares súbitas), ausencias atípicas, crisis focales y crisis tónico-clónicas generalizadas.
- Afectación Cognitiva: A partir del segundo año de vida, se hace evidente un retraso en el desarrollo psicomotor. Se observa un estancamiento en la adquisición del lenguaje y de las capacidades cognitivas, e incluso regresiones (pérdida de habilidades previamente adquiridas).
- Hiper-termosensibilidad y Fotosensibilidad: Estímulos ambientales cotidianos como los cambios bruscos de temperatura, baños con agua templada, el esfuerzo físico, los destellos de luz o ciertos patrones visuales repetitivos desencadenan crisis de forma constante.
Adolescencia y Edad Adulta (Fase de Estabilización)
- Persistencia de la Epilepsia: Aunque las crisis convulsivas prolongadas tienden a espaciarse en el tiempo o a ser más breves, la epilepsia se mantiene activa y farmacorresistente durante toda la vida.
- Marcha Agachada (Crouch Gait): Es una alteración neuromuscular muy característica que se consolida durante la pubertad. El paciente camina con una flexión persistente de rodillas y caderas, acompañada generalmente de pies planos-valgos.
- Comorbilidades Neuroconductuales: Presentan discapacidad intelectual (de moderada a severa), rasgos del Trastorno del Espectro Autista (TEA), Trastorno por Déficit de Atención e Hiperactividad (TDAH), trastornos del sueño severos y, a nivel motor, problemas de deglución, falta de coordinación (ataxia) y rasgos parkinsonianos en etapas avanzadas.
3. Criterios de Diagnóstico
El diagnóstico temprano es crucial para modificar el curso de la enfermedad y evitar daños neurológicos severos irreversibles.
- Historia Clínica: Lactante con desarrollo normal previo que debuta con crisis prolongadas (frecuentemente unilaterales o alternantes) asociadas a fiebre o vacunación antes del primer año de vida.
- Electroencefalograma (EEG): Suele ser completamente normal al inicio de la enfermedad. A partir de los 2 o 3 años, comienza a mostrar una actividad de fondo ralentizada y descargas generalizadas y multifocales de puntas y ondas.
- Resonancia Magnética (RM): Es estructuralmente normal al inicio, lo que ayuda a descartar malformaciones cerebrales u otras causas de epilepsia. Con los años, puede mostrar atrofia cortical inespecífica o esclerosis hipocampal.
- Confirmación Genética: Se realiza mediante un panel de secuenciación genética para epilepsias, buscando mutaciones o deleciones en el gen SCN1A.
4. Tratamiento y Manejo Terapéutico
El tratamiento del Síndrome de Dravet es complejo, requiere un abordaje multidisciplinar y se divide en tres pilares esenciales.
A. Contraindicación Absoluta de Fármacos
Existe un grupo de fármacos anticrisis que están estrictamente contraindicados en pacientes con Dravet. Se trata de los bloqueadores de los canales de sodio:
MEDICAMENTOS PROHIBIDOS: Carbamazepina, Oxcarbazepina, Fenitoína, Lamotrigina, Vigabatrina y Rufinamida.
Razón clínica: Al bloquear canales de sodio que ya funcionan con un rendimiento muy bajo debido a la mutación genética, estos fármacos agravan drásticamente la frecuencia de las crisis y aumentan exponencialmente el riesgo de provocar un estado de mal epiléptico potencialmente letal.
B. Algoritmo Farmacológico Actual
El tratamiento convencional busca la politerapia (combinación de varios fármacos) para reducir al máximo la frecuencia de crisis graves.
| Línea de Tratamiento | Fármacos de Elección | Observaciones Clínicas |
| Primera Línea |
Valproato de Sodio Clobazam |
Estabilizan la actividad basal. Se pautan casi siempre como terapia de inicio combinada. |
| Segunda Línea (Añadidos) |
Estiripentol Fenfluramina Cannabidiol (pureza farmacéutica) |
La Fenfluramina y el Cannabidiol de prescripción médica han transformado el pronóstico, logrando disminuciones drásticas en el número de crisis motoras y convulsivas. |
| Tratamiento de Rescate |
Midazolam (vía bucal/nasal) Diazepam (vía rectal) |
Protocolo de emergencia que los cuidadores deben aplicar en casa si una crisis supera los 5 minutos de duración. |
C. Tratamientos Dietéticos y Terapias de Apoyo
- Dieta Cetogénica: Una dieta estricta, alta en grasas y muy baja en carbohidratos, controlada por especialistas en nutrición clínica. Ayuda a modificar el metabolismo cerebral, disminuyendo la excitabilidad de las neuronas en casos de difícil control.
- Estimulación Temprana y Rehabilitación: Fisioterapia neurológica continua (fundamental para prevenir o mitigar la marcha agachada), logopedia para el desarrollo del lenguaje, y terapia ocupacional encaminada a potenciar la autonomía del paciente.
5. Pronóstico, Mortalidad y Nuevas Terapias
El Síndrome de Dravet presenta una tasa de mortalidad general superior a la de la población con otras formas de epilepsia. El mayor riesgo proviene del SUDEP (Muerte Súbita Inesperada en la Epilepsia) y de las complicaciones derivadas de los estados de mal epiléptico prolongados.
La Revolución de la Medicina de Precisión
La investigación científica está cambiando el enfoque de la enfermedad a través del desarrollo de terapias modificadoras de la enfermedad (tratamientos dirigidos a corregir la raíz genética, no solo a paliar las convulsiones):
- Oligonucleótidos Antisentido (ASO): Fármacos como el Zorevunersen (STK-001) actúan modulando la expresión del ARN mensajero. Su función es estimular la copia sana del gen SCN1A para que produzca el doble de proteínas $Na_V1.1$, compensando el fallo de la copia mutada. Los resultados en ensayos clínicos avanzados muestran reducciones drásticas y sin precedentes en la carga global de crisis.
- Terapia Génica: Se están desarrollando vectores virales modificados (AAV) diseñados para transportar y sustituir el material genético defectuoso de forma específica dentro de las interneuronas cerebrales, buscando una potencial estabilización definitiva de la patología.
últimas noticias sobre Síndrome de Dravet
Últimas noticias