Asociación MPS-LISOSOMALES España
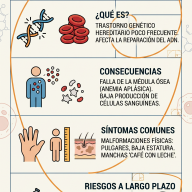
Las Mucopolisacaridosis (MPS) son un grupo de trastornos metabólicos hereditarios poco frecuentes. En términos sencillos, son problemas con el "sistema de reciclaje" de nuestras células.
Aquí tienes un desglose claro de qué son, cómo funcionan y cuáles son sus tipos.
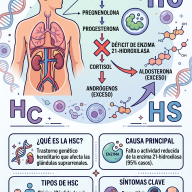
¿Qué ocurre en el cuerpo?
Normalmente, nuestras células utilizan enzimas para descomponer unas moléculas largas de azúcar llamadas glucosaminoglucanos (GAG, antes conocidos como mucopolisacáridos). Estas moléculas ayudan a construir huesos, cartílagos, tendones y piel.
En una persona con MPS:
- El cuerpo no produce suficiente cantidad de una enzima específica o la enzima no funciona correctamente.
- Los GAG no se descomponen y se acumulan en las células.
- Este exceso actúa como un "atasco" que termina dañando órganos, huesos y el sistema nervioso central.
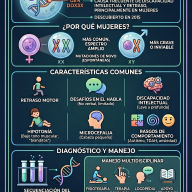
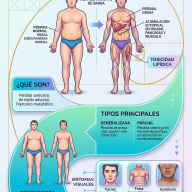
Los diferentes tipos de MPS
Aunque comparten similitudes, cada tipo se debe a la deficiencia de una enzima distinta.
| Tipo | Nombre Común | Características Principales |
| MPS I | Hurler / Scheie | Varía de leve a grave; afecta visión, articulaciones y corazón. |
| MPS II | Hunter | Ligada al cromosoma X (afecta mayormente a hombres). |
| MPS III | Sanfilippo | Principalmente afecta el cerebro y el desarrollo cognitivo. |
| MPS IV | Morquio | Afecta predominantemente el desarrollo óseo y el crecimiento. |
| MPS VI | Maroteaux-Lamy | Similar a la MPS I, pero usualmente con inteligencia normal. |
| MPS VII | Sly | Muy poco frecuente; síntomas muy variables. |
| MPS IX | Natowicz | Deficiencia de hialuronidasa; afecta las articulaciones. |
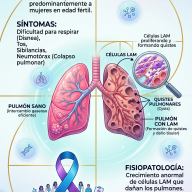
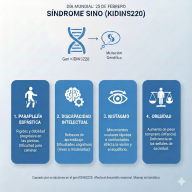
Síntomas Comunes
Dado que los GAG se acumulan en todo el cuerpo, los síntomas suelen ser multisistémicos:
- Rasgos faciales toscos: Labios gruesos, puente nasal plano y frente prominente.
- Problemas óseos: Baja estatura, rigidez articular y deformidades en la columna.
- Dificultades respiratorias: Debido a la obstrucción de las vías aéreas y agrandamiento de amígdalas/adenoides.
- Agrandamiento de órganos: Especialmente el hígado y el bazo (hepatoesplenomegalia).
- Problemas neurológicos: Pérdida de habilidades adquiridas o retraso en el desarrollo (especialmente en el tipo III).
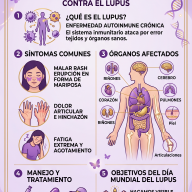
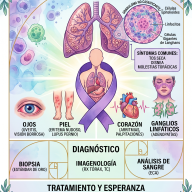
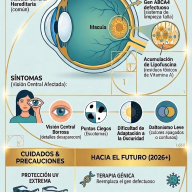
Diagnóstico y Tratamiento
El diagnóstico suele confirmarse mediante un análisis de orina (para detectar niveles altos de GAG) y se ratifica con un análisis de sangre para medir la actividad enzimática.
Nota esperanzadora: Aunque no existe una cura definitiva, la medicina ha avanzado mucho. Actualmente existen:
- Terapia de Reemplazo Enzimático (TRE): Se administra la enzima faltante por vía intravenosa.
- Trasplante de Células Madre: Útil en ciertos casos para frenar el daño neurológico.
- Terapias génicas: Actualmente en investigación para corregir el problema desde el ADN.
Es un camino complejo que requiere un equipo médico multidisciplinar (genetistas, cardiólogos, ortopedistas, etc.) para mejorar la calidad de vida del paciente.
Al ser un grupo de enfermedades, la incidencia total es la suma de sus diferentes tipos, cada uno ligado a una deficiencia enzimática específica.
- El conjunto de todos los tipos de MPS ocurre en aproximadamente 1 de cada 20,000 a 25,000 nacimientos
Tipo Gen Implicado Incidencia Estimada (Nacimientos) MPS I (Hurler/Scheie) IDUA 1 : 100,000 (grave) a 1 : 500,000 (leve) MPS II (Hunter) IDS (Ligado al X) 1 : 100,000 a 1 : 150,000 varones MPS III (Sanfilippo) SGSH, NAGLU, HGSNAT, GNS 1 : 50,000 a 1 : 280,000 (según subtipo) MPS IV (Morquio) GALNS (tipo A), GLB1 (tipo B) Aproximadamente 1 : 200,000 a 1 : 300,000 MPS VI (Maroteaux-Lamy) ARSB Muy rara (variable según región) MPS VII (Sly) GUSB Menos de 1 : 1,000,000
Casi todas las MPS son autosómicas recesivas (ambos padres deben portar el gen), la MPS II (Hunter) es la única ligada al cromosoma X, afectando casi exclusivamente a hombres.
El cribado neonatal de las Mucopolisacaridosis (MPS) es un proceso de detección precoz fundamental para identificar estas enfermedades raras antes de que aparezcan síntomas clínicos irreversibles. Aunque su inclusión en los programas de salud pública varía según el país y la región, la tendencia mundial —incluida España en 2026— es hacia su incorporación progresiva debido a la disponibilidad de tratamientos eficaces.
- Técnica: Se utiliza espectrometría de masas en tándem (MS/MS) para medir la actividad de enzimas específicas en la mancha de sangre seca.
- Confirmación: Un resultado positivo en el cribado no es un diagnóstico definitivo; requiere pruebas de seguimiento en leucocitos, análisis de glucosaminoglucanos (GAG) en orina y estudios genéticos para confirmar la mutación.
- En España, el sistema de cribado neonatal está en una fase de expansión significativa:
- Proyectos Piloto: Existen iniciativas avanzadas como el proyecto CrinGenEs (2025-2026), en el que participan 9 comunidades (incluyendo Cataluña, Madrid, Galicia y Valencia). Este proyecto utiliza cribado genómico para detectar más de 300 enfermedades raras, entre ellas diversos tipos de MPS, permitiendo un diagnóstico mucho más amplio que el bioquímico tradicional.
Importancia de la detección precoz
El éxito del tratamiento en las MPS depende críticamente de cuándo se inicia:
- Tratamientos disponibles: La Terapia de Sustitución Enzimática (TSE) y el Trasplante de Progenitores Hematopoyéticos (TPH) pueden frenar o prevenir daños graves.
- Prevención de daños irreversibles: Una vez que se manifiestan los síntomas óseos, valvulares cardíacos o el deterioro cognitivo, el tratamiento es mucho menos eficaz.
- Evitar la "odisea diagnóstica": Sin cribado, las familias pueden tardar años en obtener un diagnóstico, perdiendo una ventana de oportunidad terapéutica vital.
Últimas noticias