Asociación Española de Pacientes con Enfermedad Asociada a IgG4
Este es un desglose detallado y estructurado sobre la Enfermedad Relacionada con IgG4 (ER-IgG4). Este informe aborda desde sus mecanismos celulares latentes hasta los criterios diagnósticos internacionales y las innovaciones terapéuticas más recientes.
1. Naturaleza y Contexto Histórico
La ER-IgG4 es una patología fibroinflamatoria sistémica mediada por inmunidad que puede afectar prácticamente a cualquier órgano. Durante el siglo XX, los médicos describieron síndromes aislados en diversos órganos sin saber que compartían la misma raíz.
Todo cambió a principios de los años 2000 en Japón, cuando se descubrió que los pacientes con pancreatitis esclerosante (ahora Pancreatitis Autoinmune Tipo 1) tenían niveles llamativamente altos de anticuerpos IgG4 en la sangre y un infiltrado celular idéntico en sus tejidos. Poco después, se evidenció que este mismo patrón ocurría en glándulas salivales, riñones y retroperitoneo, unificando el mapa de la enfermedad.
2. Fisiopatología: ¿Qué ocurre a nivel celular?
Una de las grandes paradojas de esta enfermedad es que la molécula de IgG4 en sí misma no parece ser la causante del daño. En condiciones normales, la IgG4 es un anticuerpo bloqueador y antiinflamatorio. En esta patología, su elevación masiva parece ser un intento del cuerpo por "frenar" una inflamación descontrolada dirigida por otras células.
El verdadero motor de la enfermedad es una interacción celular anómala:
- Linfocitos T Cooperadores (Tfh y CD4+ citotóxicos): Estas células se activan de forma aberrante y secretan grandes cantidades de citoquinas inflamatorias y profibróticas (como el factor de crecimiento transformante beta o TGF-$\beta$, y las interleucinas IL-4 e IL-10).
- Activación de Fibroblastos: Estas citoquinas actúan como una orden directa para que los fibroblastos produzcan colágeno de forma masiva y desorganizada, originando la característica fibrosis estoriforme (tejido cicatrizal con un patrón en forma de estera tejida).
- Reclutamiento de Células B: Al mismo tiempo, se reclutan y expanden linajes de células B y células plasmáticas que se dedican a fabricar anticuerpos IgG4 de manera masiva, inundando el tejido afectado.
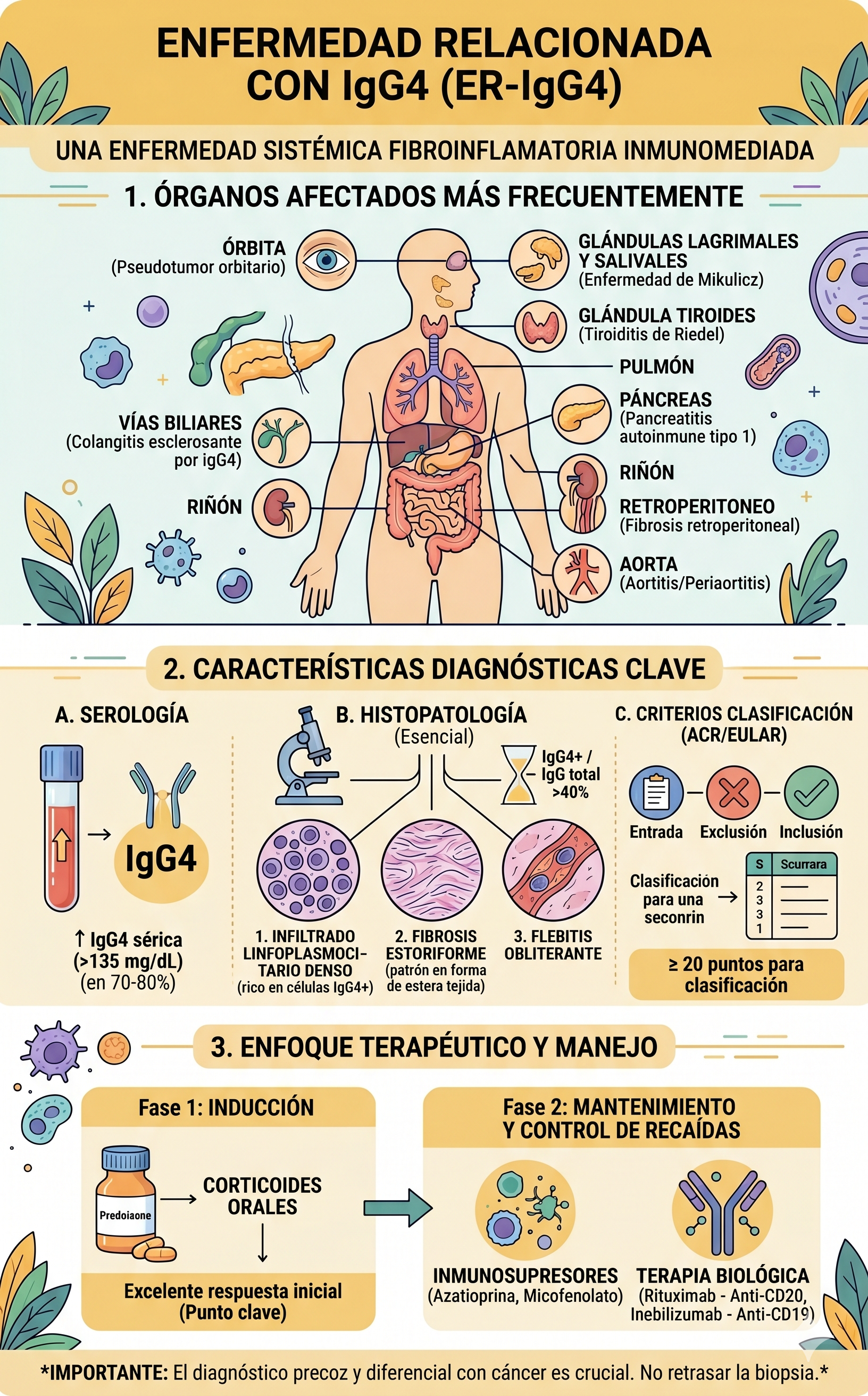
3. Criterios Diagnósticos Internacionales (ACR/EULAR)
Debido a que la ER-IgG4 es una excelente "imitadora" de tumores malignos, el Colegio Americano de Reumatología (ACR) y la Liga Europea Contra el Reumatismo (EULAR) establecieron una guía estricta en tres pasos para evitar errores de diagnóstico:
Paso 1: Criterio de Entrada
El paciente debe presentar un compromiso clínico o radiológico característico en al menos uno de los órganos diana (páncreas, glándulas salivales, órbitas, pulmón, riñón, retroperitoneo, aorta, etc.).
Paso 2: Criterios de Exclusión
Se deben descartar rigurosamente otras patologías. Si el paciente presenta marcadores de cáncer evidentes, infecciones activas (como tuberculosis u hongos), o datos de otras enfermedades autoinmunes específicas (como sarcoidosis o vasculitis ANCA), el diagnóstico de ER-IgG4 queda excluido.
Paso 3: Criterios de Inclusión (Puntuación)
Se evalúan 8 dominios que combinan hallazgos clínicos, analíticos, radiológicos e histopatológicos. Cada hallazgo suma puntos. Se requiere una puntuación mínima de 20 puntos para confirmar la clasificación.
| Dominio | Hallazgo de Alto Puntaje |
| Histopatología | Presencia de los 3 rasgos clave (infiltrado linfoplasmocitario, fibrosis estoriforme y flebitis obliterante). |
| Inmunotinción | Relación de células plasmáticas IgG4+ / IgG totales mayor al 40% en el tejido. |
| Serología | Niveles de IgG4 sérica superiores a 5 veces el límite normal. |
| Radiología | Agrandamiento difuso del páncreas ("páncreas en salchicha") o fibrosis retroperitoneal que envuelve la aorta. |
4. Diagnóstico Diferencial: El Gran Reto
El principal peligro de la ER-IgG4 es el retraso diagnóstico o la confusión con procesos oncológicos. Al presentarse como masas densas e indoloras, muchos pacientes han sido sometidos históricamente a cirugías agresivas (como la operación de Whipple para remover el páncreas) bajo la sospecha de un cáncer que terminó siendo una inflamación benigna por IgG4.
Puntos clave de diferenciación: A diferencia de las infecciones o las vasculitis sistémicas, la ER-IgG4 rara vez produce fiebre alta o elevaciones extremas de la proteína C reactiva (PCR). Su avance es lento, insidioso y de aspecto tumoral.
5. Estrategia de Tratamiento y Avances Recientes
El manejo clínico se divide en dos fases críticas, donde la inmunoterapia dirigida ha ganado un terreno histórico:
A. Inducción de la Remisión
Los glucocorticoides por vía oral (habitualmente prednisona a dosis de 0.6 mg/kg/día) siguen siendo la primera línea. La respuesta suele ser dramática: en cuestión de dos a cuatro semanas, las masas inflamatorias disminuyen drásticamente de tamaño. Si una supuesta masa por IgG4 no responde en absoluto a los esteroides, obliga al médico a replantearse el diagnóstico inmediatamente.
B. Mantenimiento y Control de Recaídas
El gran problema de esta enfermedad es que, al retirar por completo los esteroides, entre el 30% y el 50% de los pacientes sufre una recaída. Para evitar depender crónicamente de los corticoides (y su conocida toxicidad), se recurre a terapias biológicas:
- Rituximab: Este anticuerpo monoclonal destruye selectivamente las células B que expresan el marcador CD20, cortando el suministro de nuevas células plasmáticas inflamatorias. Ha sido el pilar de rescate más utilizado en la última década.
- Inebilizumab (Aprobación FDA): Un hito terapéutico clave ocurrió con la aprobación formal de este anticuerpo monoclonal dirigido contra el receptor CD19. Al atacar un espectro más amplio de células de linaje B (incluyendo algunas células plasmáticas tempranas que el Rituximab no logra erradicar), ha demostrado reducir drásticamente el riesgo de brotes en pacientes con enfermedad multiorgánica activa, marcando una nueva era de precisión en el tratamiento de la ER-IgG4.
últimas noticias sobre Enfermedad asociada a IgG4
Últimas noticias




