Asociación Española de Enfermos Pompe
La enfermedad de Pompe (también conocida como glucogenosis tipo II) es un trastorno genético poco frecuente, debilitante y, a menudo, mortal. Pertenece al grupo de las enfermedades por depósito lisosomal.
Aquí tienes un resumen detallado de lo que necesitas saber:
¿Qué es y qué la causa?
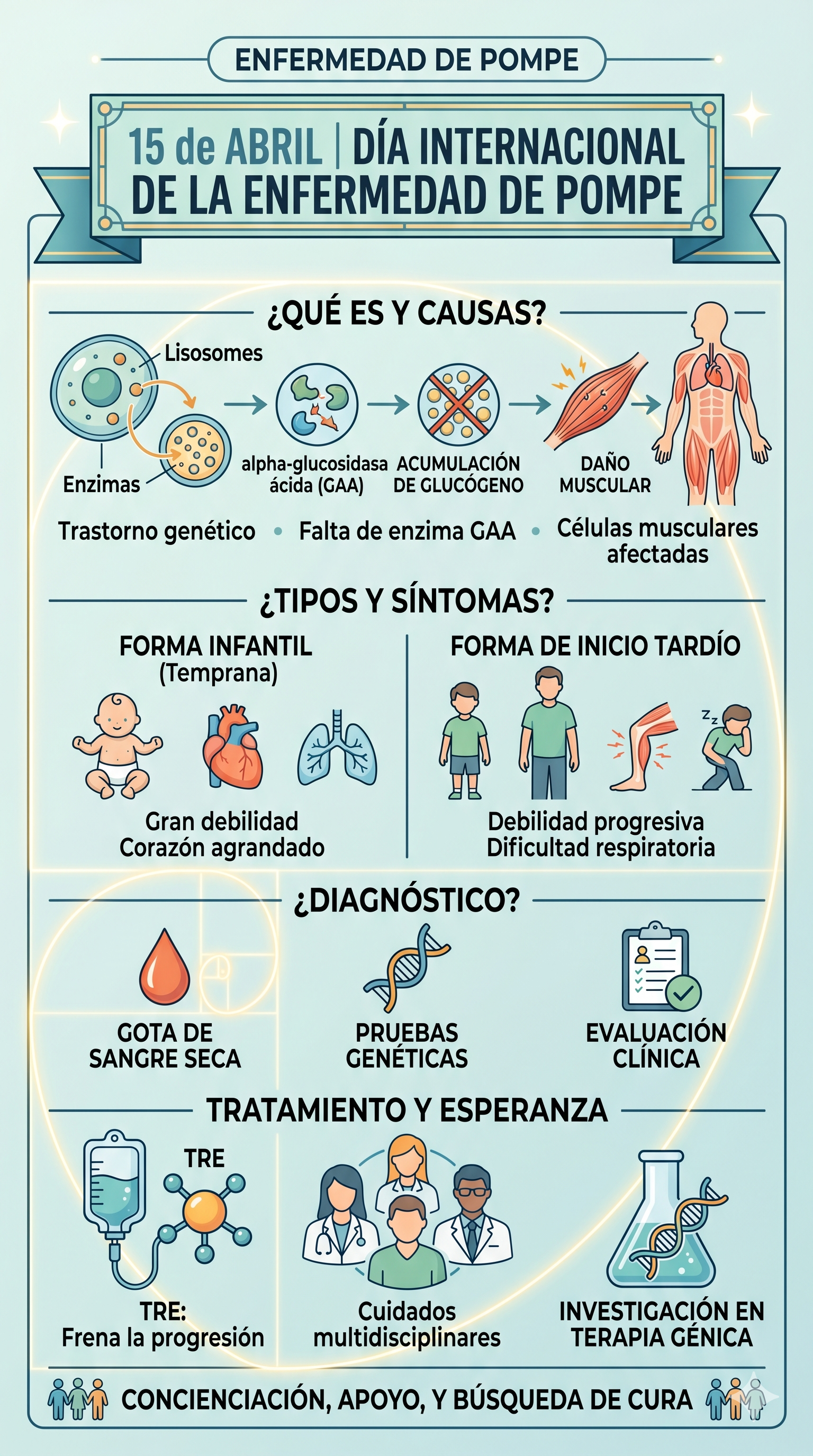
Se produce por una deficiencia o ausencia de la enzima alfa-glucosidasa ácida (GAA).
- El mecanismo: Normalmente, esta enzima descompone el glucógeno (azúcar complejo) en glucosa dentro de los lisosomas de las células.
- El problema: Al no haber suficiente enzima, el glucógeno se acumula masivamente, dañando principalmente las células musculares (esqueléticas y cardíacas).
- Herencia: Es una enfermedad autosómica recesiva, lo que significa que el paciente debe heredar una copia defectuosa del gen de cada progenitor.
Tipos de la enfermedad
La gravedad depende de cuánta actividad enzimática mantenga el cuerpo:
1. Forma infantil (IOPD)
Es la más grave y aparece en los primeros meses de vida.
- Síntomas: Gran debilidad muscular (bebé "blando" o hipotónico), dificultad para succionar y tragar, y cardiomegalia (corazón extremadamente agrandado).
- Pronóstico: Sin tratamiento, suele ser fatal antes del primer año de vida debido a insuficiencia cardíaca o respiratoria.
2. Forma de inicio tardío (LOPD)
Puede aparecer en la infancia tardía, adolescencia o edad adulta.
- Síntomas: Debilidad muscular progresiva (especialmente en piernas y tronco) y dificultades respiratorias por debilidad del diafragma. El corazón no suele verse afectado.
- Progreso: Es más lenta, pero puede llevar a la necesidad de silla de ruedas o soporte ventilatorio.
Diagnóstico
El diagnóstico temprano es vital para mejorar el pronóstico. Se realiza mediante:
- Gota de sangre seca (DBS): Una prueba rápida para medir la actividad de la enzima GAA.
- Pruebas genéticas: Para confirmar mutaciones en el gen GAA.
- Biopsia muscular: (Menos común hoy en día) para ver la acumulación de glucógeno.
Tratamiento actual
Aunque no existe una cura definitiva, el manejo ha cambiado radicalmente en los últimos años:
- Terapia de Reemplazo Enzimático (TRE): Consiste en administrar por vía intravenosa la enzima que le falta al paciente (como la alglucosidasa alfa o la avalglucosidasa alfa). Esto ayuda a reducir la acumulación de glucógeno y frena la progresión de la enfermedad.
- Cuidado multidisciplinar: Apoyo de fisioterapeutas, neumólogos, cardiólogos y nutricionistas (se suele recomendar una dieta alta en proteínas).
- Terapia Génica: Actualmente hay investigaciones avanzadas para intentar que el propio cuerpo del paciente produzca la enzima de forma permanente.
Nota importante: Si sospechas de síntomas relacionados o tienes antecedentes familiares, es crucial acudir a un neurólogo o genetista especializado en enfermedades metabólicas.
La enfermedad de Pompe
Es una enfermedad neurodegenerativa e incapacitante.
Una enfermedad rara que puede afectar a personas de cualquier edad y origen étnico, bebés, niños y adultos. Es de origen genético y recesiva, lo que significa que se transmite en las familias de padres a hijos cuando ambos progenitores son enfermos o portadores y es también conocido como un trastorno metabólico.
En la actualidad afecta a unas 120 personas en España, pero hay muchos más enfermos que son asintomáticos todavía y no están diagnosticados. Puede provocar diversos problemas de salud graves, el primer síntoma más frecuente es la debilidad muscular que empeora con el tiempo y acaba afectando también al sistema respiratorio y cardiaco.
La enfermedad de Pompe puede recibir los siguientes nombres, pero todos ellos se refieren exactamente a la misma enfermedad:
- Deficiencia de maltasa ácida
- Enfermedad de acumulación Formación de glucógeno de tipo II
- Glucogenosis de tipo II
- Deficiencia de alfa glucosidasa ácida
- Deficiencia de alfa glucosidasa lisosómica
Últimas noticias