Asociación para la Información e Investigación de la Hipomagnesemia Familiar
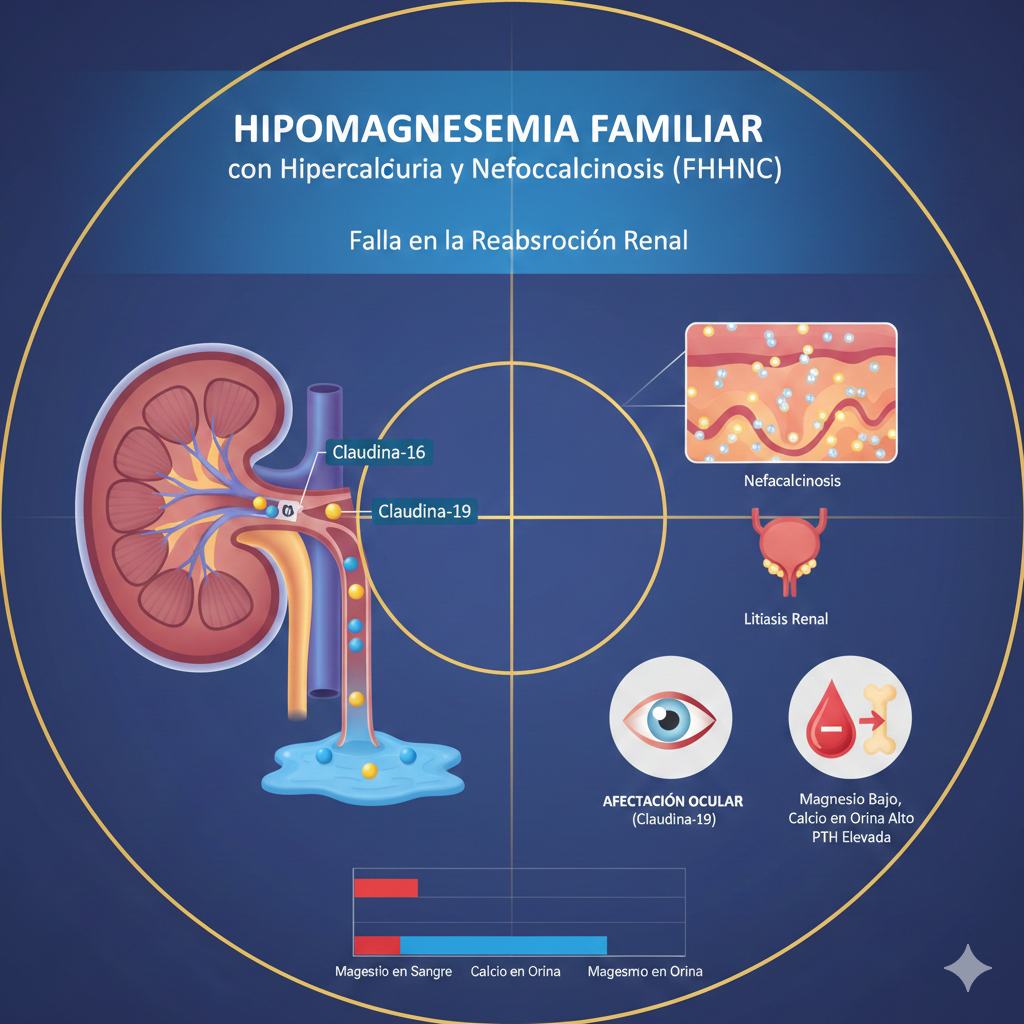
La Hipomagnesemia Familiar con Hipercalciuria y Nefrocalcinosis (FHHNC) es una enfermedad renal genética poco común, pero de gran importancia clínica. Se caracteriza por una pérdida excesiva de magnesio y calcio a través de la orina, lo que eventualmente daña el tejido renal.
Aquí te detallo los aspectos clave para entender este trastorno:
1. ¿Qué ocurre en el riñón?
Normalmente, el magnesio y el calcio se reabsorben en una parte específica de la nefrona llamada el asa de Henle (rama ascendente gruesa).
En la FHHNC, existe una mutación en las proteínas encargadas de "abrir la puerta" para que estos minerales regresen a la sangre. Estas proteínas son la Claudina-16 y la Claudina-19. Al no poder reabsorberse, el magnesio y el calcio se quedan en la orina.
2. Manifestaciones Clínicas
Los síntomas suelen aparecer en la infancia o adolescencia y se presentan como:
- Poliuria y polidipsia: Necesidad de orinar mucho y beber mucha agua.
- Infecciones urinarias recurrentes: Debido a la formación de cristales.
- Nefrocalcinosis: Es la acumulación de depósitos de calcio en el parénquima renal, visible en ecografías.
- Litiasis renal: Formación de piedras (cálculos).
- Falla renal progresiva: El daño constante puede llevar a la enfermedad renal crónica terminal.
3. Hallazgos de Laboratorio
Si se sospecha de esta condición, los resultados típicos muestran:
| Parámetro | Resultado |
| Magnesio en sangre | Bajo (Hipomagnesemia) |
| Magnesio en orina | Elevado (Hipermagnesiuria) |
| Calcio en orina | Elevado (Hipercalciuria) |
| Hormona Paratiroidea (PTH) | Elevada (Hiperparatiroidismo secundario) |
Nota importante: A diferencia de otros trastornos, el calcio en la sangre suele mantenerse en niveles normales, pero a costa de "sacarlo" de los huesos debido a la PTH elevada.
4. Diferenciación Genética
Existen dos tipos principales según el gen afectado:
- Tipo 1 (Claudina-16): Afectación puramente renal.
- Tipo 2 (Claudina-19): Además de los problemas renales, los pacientes suelen presentar defectos oculares graves, como miopía severa, nistagmo o coloboma.
Para profundizar en la diferencia entre estas dos proteínas, es necesario entender que ambas forman parte de las uniones estrechas (tight junctions) en el riñón, pero la Claudina-19 tiene una expresión más amplia en el cuerpo.
Aquí tienes las diferencias clave divididas por su impacto clínico y biológico:
1. Ubicación y Función Biológica
- Claudina-16 (Paracelina-1): Se expresa casi exclusivamente en el riñón, específicamente en la rama ascendente gruesa del asa de Henle. Su función es crear un canal que permite el paso selectivo de cationes (magnesio y calcio) desde la orina hacia la sangre.
- Claudina-19: Además de estar en el riñón (donde interactúa directamente con la Claudina-16 para formar los canales de reabsorción), se expresa fuertemente en el epitelio pigmentario de la retina y en las vainas de mielina del sistema nervioso periférico.
2. Manifestaciones Extra-renales (La gran diferencia)
Esta es la forma más sencilla de distinguirlas clínicamente:
- Mutaciones en CLDN16 (FHHNC Tipo 1): El paciente presenta síntomas puramente renales (nefrocalcinosis, pérdida de magnesio, piedras en el riñón y fallo renal progresivo). No suele haber afectación en otros órganos.
- Mutaciones en CLDN19 (FHHNC Tipo 2): Los pacientes presentan los mismos problemas renales severos, pero siempre se asocian con anomalías oculares graves. Esto se conoce como el síndrome de hipomagnesemia renal con compromiso ocular.
3. Fenotipo Ocular (Exclusivo de Claudina-19)
Si un paciente con hipomagnesemia y nefrocalcinosis presenta alguno de los siguientes síntomas, la causa es casi con seguridad una mutación en la Claudina-19:
- Miopía magna (severa): Generalmente presente desde el nacimiento.
- Nistagmo: Movimiento involuntario e incontrolable de los ojos.
- Coloboma coriorretiniano: Un "agujero" o falta de tejido en las capas internas del ojo.
- Estrabismo y astigmatismo marcado.
4. Gravedad y Progresión
Aunque ambos tipos son graves, diversos estudios sugieren algunas diferencias en la evolución:
- Progresión a Insuficiencia Renal: En ambos casos, el deterioro de la función renal es rápido, pero algunos registros indican que los pacientes con mutaciones en Claudina-19 podrían llegar a la enfermedad renal crónica terminal (necesidad de diálisis o trasplante) a una edad ligeramente más temprana que los de Claudina-16.
- Niveles de Magnesio: La hipomagnesemia tiende a ser un poco más profunda en el Tipo 2 (CLDN19).
Resumen comparativo
| Característica | Claudina-16 (Tipo 1) | Claudina-19 (Tipo 2) |
| Afectación Renal | Grave (Nefrocalcinosis) | Grave (Nefrocalcinosis) |
| Afectación Ocular | Ausente | Presente (Miopía, Coloboma) |
| Localización | Solo Riñón | Riñón y Retina |
| Interacción | Necesita a la CLDN19 | Necesita a la CLDN16 |
En conclusión: Si ves a un niño con pérdida de magnesio y calcio en la orina que además usa gafas con mucha graduación o tiene problemas de visión estructurales, el diagnóstico genético más probable es una mutación en el gen CLDN19.
5. Tratamiento y Manejo
El objetivo no es "curar" la mutación (que no es posible actualmente), sino minimizar el daño:
- Suplementos de Magnesio: Dosis altas por vía oral (aunque pueden causar diarrea).
- Diuréticos Tiazídicos: A veces se usan para reducir la excreción de calcio en la orina.
- Citrato de potasio: Para prevenir la formación de cristales de oxalato de calcio.
- Seguimiento estrecho: Monitoreo de la función renal para retrasar la necesidad de diálisis o trasplante.